Shredded Vegetarian Beef,Vegetarian Shredded Beef,Instant Shredded Vegetarian Beef,Frozen Shredded Vegetarian Beef Guangyun Agricultural Biotechnology (Jiangsu) Co., Ltd , https://www.7-mushrooms.com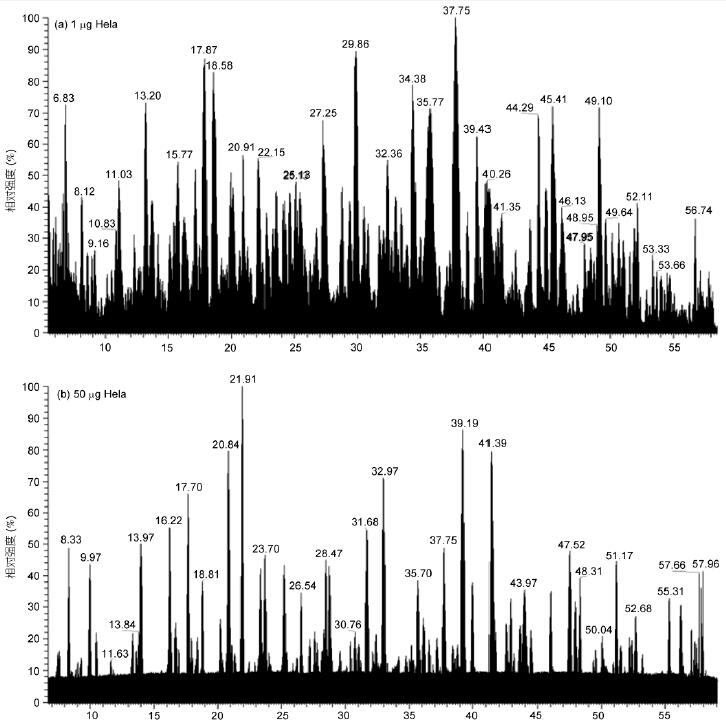

One-hour proteome: rapid proteomics identification based on Q-OT-qIT mass spectrometry
Zhang Weiwei, Gu Peiming†, Jiang Yan*, Ming Hong, Chen Wei
Thermo Fisher Scientific (China) Co., Ltd., Shanghai 201206
åŒç‰ Equal contribution
* Contact, E-mail: zheng. Jiang@thermofisher. Com
Abstract With the realization of the “one-hour yeast proteomeâ€, it is possible to achieve proteomic deep coverage under a short gradient. This article uses the new Q-OT-qIT three-in-one mass spectrometry system for "one-hour proteome" analysis and optimization. 50 min effective gradient, 20860 and 14100 peptides were identified from 1 μg and 50 ng HeLa whole protein in a single experiment, corresponding to 3865 and 2877 non-redundant proteins, and the current literature reports at least 2~3 h . At the same time, this paper examines the effects of parameters such as Q-OT-qIT fragmentation mode, detection method, maximum injection time and automatic gain control on rapid proteomic analysis, and proves the complementarity between different acquisition methods. The impact of the identification results. In addition, the parallel operation principle and scanning combination mode of Q-OT-qIT are discussed, which lays a foundation for rapid analysis of proteomes for different experimental purposes and sample types.
Key words proteomics rapid identification depth coverage electrostatic field orbital trap Q-OT-qIT mass spectrometry
Proteomic in-depth proteome has become a trend in proteomics research, providing important information for the study of signaling pathways, molecular targets and biomarkers through large-scale identification and mining of very low-abundance proteins. . However, due to the complexity of whole protein samples, proteomic depth identification requires high chromatographic separation, usually requiring two-dimensional separation (strong anion exchange, high pH reverse phase, etc.), or using one-dimensional long gradient separation to reduce high Abundance protein interference [1,2]. However, this consumes a lot of time and reproducibility is not good, which becomes the bottleneck of proteome deep coverage research.
The electrostatic field orbitrap mass analyzer has ultra-high resolution and ultra-high sensitivity, which effectively promotes the development of proteomics [3, 4]. The new Q-OT-qIT (Orbitrap Fusion) mass spectrometry system combines quadrupole (Q), electrostatic field orbitrap (OT) and linear quadrupole ion trap (qIT) for the first time. As one, using dynamic scanning management technology, three kinds of quality analyzers work simultaneously and cooperate with each other, and the first-level and second-level scanning are simultaneously performed to maximize the analysis efficiency [5]. Hebert et al. [6] used Q-OT-qIT mass spectrometry, 5 replicate experiments, and identified about 4000 yeast proteins in an effective gradient of 70 min. The same results, the current literature reports at least 4 h, analysis efficiency (unit time protein The number of identifications has increased by more than 4 times [7, 8]. Based on this, Hebert et al. [6] first proposed the concept of "one-hour yeast proteome", that is, a short gradient of about 1 h to achieve deep coverage of yeast proteome. Proteomics has entered a new era.
In this paper, the Q-OT-qIT mass spectrometry system was used to rapidly identify the whole protein of HeLa and yeast cells by short gradient, and the "one hour proteome" based on Q-OT-qIT mass spectrometry was further investigated and optimized. The results showed that a single experiment, 50-minute effective gradient, identified 20860 and 14100 peptides from 1 μg and 50 ng HeLa, corresponding to 3865 and 2877 non-redundant proteins, and the analysis efficiency was improved by at least 2 compared with the current report. ~3 times [9]. This paper also compares and optimizes a variety of scan combinations and parameter settings, providing the most suitable acquisition mode for each type of experiment.
1 Materials and methods
1.1 Sample preparation
Take appropriate amount of HeLa and yeast cell pellets, add 7 mol / L urea, 2 mol / L thiourea, 1 mmol / L phenylmethanesulfonyl fluoride (PMSF) and 50 mmol / L dithiothreitol (DL-dithiothreitol, DTT) protein extract (added protease inhibitor before use), sonicated in an ultrasonic cell disrupter for 3 × 5 s. After standing on ice for 20 min, centrifuge at 30 ° C for 30 min (15000 × g ) and take Qing, Bradford method to determine the total protein concentration.
DTT was added to the protein extract to a final concentration of 10 mmol/L, and shaken at 56 ° C for 30 min. Then add iodoacetamide (IAM) to a final concentration of 50 mmol/L, and shake at room temperature for 40 min. After the reaction, 6 times volume of pre-cooled acetone was added, and the mixture was placed at 20 ° C for 3 h, and then centrifuged at 4 ° C for 30 min (15000 × g ) to discard the supernatant. The precipitate was redissolved in 50 mmol/L NH4HCO3 buffer at a concentration of 10 μg/μL. Solution, and add enough trypsin (trypsin), 37 °C enzymatic hydrolysis overnight. After enzymatic hydrolysis, the whole proteolytic solution was diluted to a final concentration of 0.5 μg / μL.
1.2 Nanoflow liquid chromatography
Chromatograph: Nanoflow Ultra Performance Liquid Chromatography EASY-nLC 1000 (Thermo, USA); Column: Nanoflow C18EASY-Spray column (2 μm, 100 Å, 75 μm × 25 cm / 50 cm), flow rate 250nL / min Self-made nano-flow C18 column (3 μm, 100 Å, 75 μm × 15 cm), flow rate 300nL/min; mobile phase A: 0.1% formic acid-water solution; mobile phase B: 0.1% formic acid-acetonitrile solution.
50min effective gradient: 0~2min, 3%~5% B; 2~42min, 5%~22% B; 42~52min, 22%~30% B; 52~55min, 30%~90% B; 55~ 60min, 90% B. 70min effective gradient: 0~5 min, 3%~8% B; 5~60min, 8%~20% B; 60~75min, 20%~30% B; 75~80min, 30% ~90% B; 80~90min, 90% B.
1.3 Mass Spectrometry
Mass spectrometer: Orbitrap Fusion (Q-OT-qIT) high resolution mass spectrometer (ThermoFisher, USA); ion source: EASY-Spray or Nanospray Flex nanoliter electrospray source; spray voltage: 2.2 kV; ion transfer tube temperature: 275 °C ; RF-lens: 60%.
Scan mode: Top-speed; Cycle time: 3 s; Dynamic exclusion: 60 s.
Primary scan: mass analyzer, orbitrap; scan range, m/z 350~1800; resolution, 120k; maximum injection time, 50 ms; automatic gain control (AGC): 2e5.
Secondary IT scan: ion selection, quadrupole; selection window, 2 amu; mass analyzer, ion trap (IT); scan speed, Rapid; maximum injection time, 35 ms; AGC: 1e4; , HCD; fragmentation energy, 35%.
Secondary OT scan: ion selection, quadrupole; selection window, 2 amu; mass analyzer, orbital well; resolution, 15k; maximum injection time, 35 ms; AGC, 1e5; fragmentation mode, HCD; , 35%.
1.4 Data Analysis
Data was identified using Proteome Discoverer 1.4 software search engine: SEQUEST HT; Database: Uniprot Human and Yeast Protein Database; Protease: Trypsin (full); Maximum Missing Site: 2; Mass Ion Mass Accuracy: 10 ppm; Ion mass accuracy: 0.6 Da(qIT)/0.02 Da(OT); Fixed modification: Carbamidomethyl(C+57.021 Da); Variable modification: acetyl (N-terminus+42.011 Da), deamidated(N, Q+0.984 Da) , oxidation (M+15.995 Da). Search results using Percolator to calculate the q value for the card value. At the same time, using Preview software to analyze the data quality.
2 Results and analysis
2.1 Q-OT-qIT short gradient analysis of HeLa and yeast proteome
Experiments were performed on 50 μL effective gradients of 1 μg and 50 ng HeLa, and 70 μL effective gradient collection of 1 μg yeast whole protein, using OT-HCD-IT scanning mode, first-order orbitrap, secondary ion trap scanning, HCD high-energy fragmentation The results showed that the proteolysis products were well separated and the mass spectrometry response was good at the peak time (Fig. 1). Even at a very low sample size of 50 ng, good separation and mass spectrometry responses were obtained (Fig. 1B).
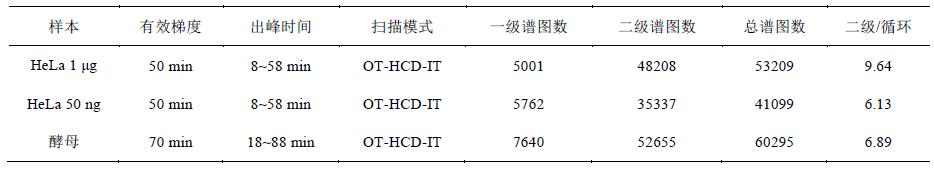
A total of 53209 spectra were obtained from a 1 μg HeLa sample in an effective gradient of 50 min, with an average of 1.7 primary spectra per second, 16.1 secondary spectra, and an average of 9.6 secondary spectra per cycle. Yeast samples were within a 70-minute effective gradient. A total of 60,295 spectra were obtained, with an average of 1.8 primary spectra per second and 12.5 secondary spectra, with an average of 6.9 secondary spectra per cycle (Table 1). HeLa cells are more complex than yeast, and there are more proteins. Therefore, more secondary spectra are obtained. In addition, at a very low sample size of 50 ng HeLa, the total number of spectra is also 41099.
Figure 1 LC-MS/MS total ion chromatogram of HeLa and yeast proteolytic products
A: 1 μg HeLa; B: 50 ng HeLa; C: 1 μg yeast
Table 1 Analysis of collected data within the effective gradient
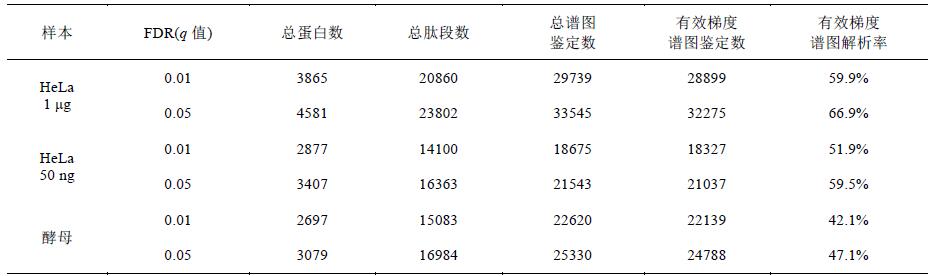
Table 2 Results of whole protein identification of HeLa and yeast samples
2.2 Rapid identification results of HeLa and yeast proteome
Data were identified from the 1 μg and 50 ng HeLa samples to 20860 and 14100 peptides ( q value 0.01), corresponding to 3865 and 2877 non-redundant proteins, respectively, by the SEQUEST HT search and Percolator card values ​​(Table 2). The same number of identification, the current report requires at least 2 ~ 3h [9]. In addition, yeast samples identified a total of 15083 peptides ( q value of 0.01), corresponding to 2697 non-redundant proteins (Table 2).
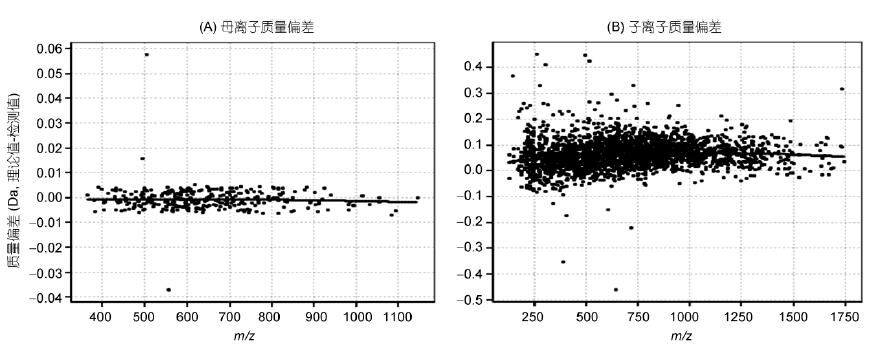
The HeLa data was further analyzed using Specview software for spectral quality accuracy (Fig. 2). The results showed that the precursor ion ( m/z ) mass accuracy averaged 0 ppm and the daughter ion mass accuracy ( m/z ) averaged 0.075 Da. The deviation is kept within 5 ppm, with high quality precision and long-term stable quality axis. The ion trap quality deviation is kept within ±0.2 Da, and the quality accuracy is good, which ensures the data quality and the reliability of the result.
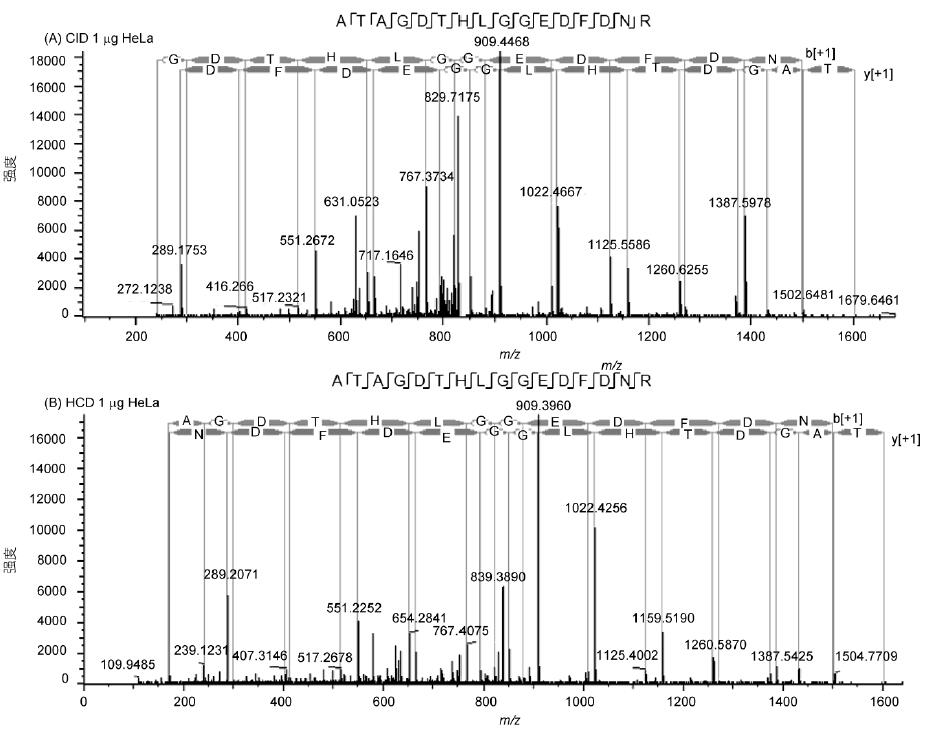
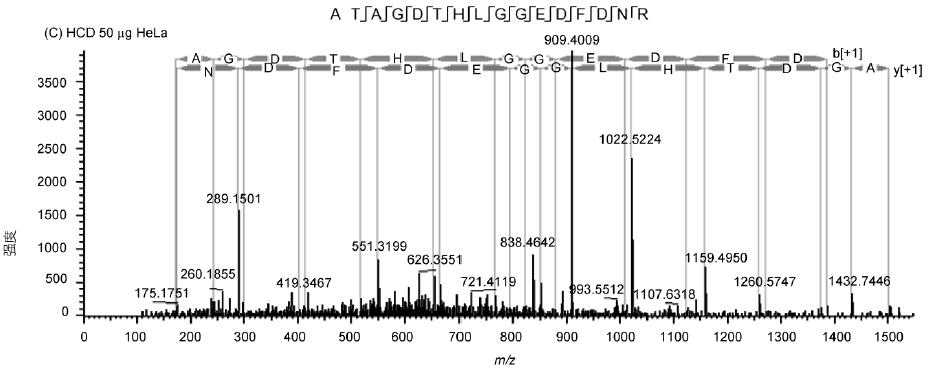
The spectrum of the peptide ATAGDTHLGGEDFDNR shows the fragmentation effect and sensitivity of the Q-OT-qIT mass spectrum (Fig. 3). The spectrum has rich fragmentation information, good matching and high sensitivity. In addition, HCD and CID are available. Fragmentation mode is quite effective and has its own characteristics: HCD avoids one-third of CID effect, low molecular weight end information is not lost, and speed is 5% faster than CID; CID sensitivity is higher, and low abundance peptide fragments are broken. The cracking effect is more obvious.
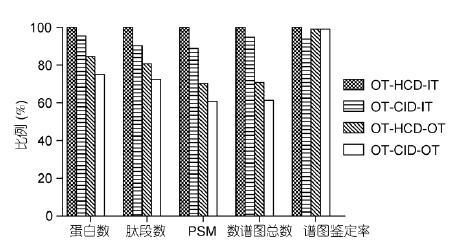
2.3 Comparison and analysis of different fragmentation modes and detection methods
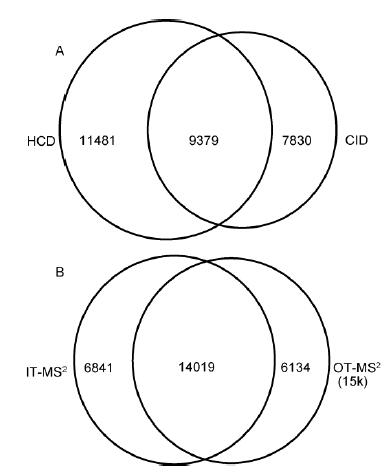
In view of the obvious difference in the identification effect of different modes, the experiment deeply investigated the influence of HCD/CID fragmentation mode and the orbitrap/ion trap secondary detection method on the identification results. Firstly, HeLa was analyzed in OT-HCD-IT, OT-CID- The results of the identification of IT and OT-HCD-OT3 models were compared. The results showed that the peptides identified by HCD and CID accounted for 32.7%, and only 67.3% were identified by one fragmentation mode; OT/ At the same time, IT detected 51.9% of peptides, and only 48.1% of them were detected by one mass analyzer (Fig. 4). CID has one-third effect, resulting in low-molecular-weight quality discrimination, and HCD overcomes low-quality discrimination. , but the sensitivity is slightly lower than CID; the ion trap has higher scanning speed and sensitivity, and the high resolution and high quality accuracy of the orbitrap effectively eliminates interference from complex sample matrices. Therefore, the two fragmentation modes and detection methods have Certain complementarity.
The experiment further used OT-HCD-IT, OT-CID-IT, OT-HCD-OT and OT-CID-OT scanning modes to perform 60-minute effective gradient collection on yeast samples, and compared the differences of different scanning modes in detail. The secondary IT uses Rapid scanning and the OT uses 30k resolution. The identification results are normalized to 100% by OT-HCD-IT results (Fig. 5). The results show that the spectrum obtained is the fastest because of OT-HCD-IT. The number of graphs and identification results is the most; CID is 5% slower than HCD, so the identification result is 5% less; OT-HCD-OT is two-level high resolution, and the first and second levels cannot be operated at the same time. The number of spectra and the number of protein identifications are respectively 70% and 85% of the secondary IT scan; OT-CID-OT is the slowest, and the number of spectra and protein identification are 60% and 75% of OT-HCD-IT, respectively. In addition, the spectra of the four scan modes The resolution rates are not much different, indicating that different scan modes have little effect on data quality.
2.4 Ion maximum injection time and the effect of automatic gain control (AGC) on the results
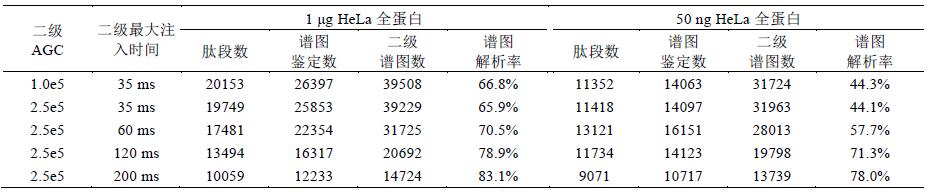
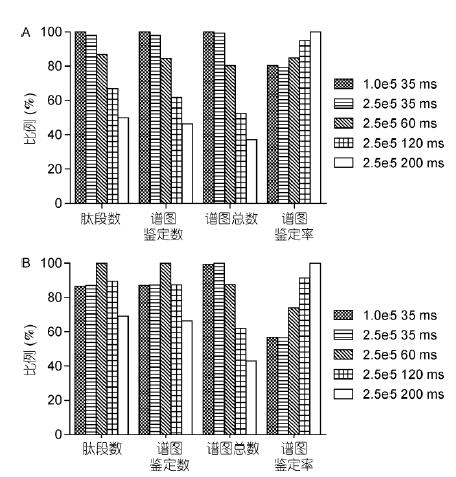
The use of C-trap to control the injection time and number of injections (automatic gain control) is a feature of the orbitrap mass analyzer. The number of ions entering the orbital trap has a great influence on the detection results. The appropriate number of ions not only avoids the space charge effect, It can also significantly improve the spectral quality, dynamic linear range and detection line. Experiments in the OT-HCD-OT mode to set different parameters for HeLa samples (secondary OT with 15k resolution), further explore different maximum injection time and The effect of AGC settings on the results (Table 2).
The identification results show that when the sample concentration is high (1 μg), the number of ions is sufficient, and the smaller injection time and AGC can obtain good results; increasing the injection time and AGC will reduce the scanning speed and reduce the number of spectra. The parameter is set to 1.0e5, and the number of identifications obtained is the highest at 35 ms (Fig. 6A). When the sample concentration is very low (50 ng HeLa), it is necessary to increase the injection time and AGC to inject more ions, improve sensitivity and spectrum. Quality; however, if the injection time is too long (120 and 200ms), the number of spectra is too small, which will result in a decrease in the identification result. Therefore, the parameter setting of 2.0e5, 60 ms obtains the most identification number (Fig. 6B). In addition, injection The longer the time, the greater the number of ions, the higher the spectral quality. Therefore, the number of ion implantation is proportional to the resolution of the spectrum.
Figure 2 Data quality accuracy analysis
A: primary quality accuracy; B: secondary quality accuracy
Table 3 Different maximum injection time and identification results of HeLa samples under AGC
Figure 3 Secondary spectrum and matching information of ATAGDTHLGGEDFDNR peptide in HeLa sample
A: 1 μg sample size, CID fragmentation; B: 1 μg sample size, HCD fragmentation; C: 50 ng sample size, HCD fragmentation
Figure 4 Comparison of the identification results of HeLa sample peptides
A: Comparison of HCD and CID fragmentation modes; B: Comparison of secondary detection of ion traps and orbitraps
Figure 5 Comparison of different scan mode data
Normalized with OT-HCD-IT as 100%
Figure 6 Comparison of different maximum ion implantation times and AGC results
A: 1 μg HeLa results comparison; B: 50 ng HeLa results comparison. The highest value for each item is 100%
3 Discussion
With the realization of the full coverage of the "one-hour yeast proteome", proteomic deep coverage research has ushered in a new era [6]. In complex sample proteome research, traditional methods require two-dimensional separation and grading to simplify the sample, Full separation, which not only requires laborious operation, but also consumes a lot of time. The Q-OT-qIT mass spectrometry technology makes it possible to realize the depth identification of complex samples under one-dimensional short gradient of about 60 min.
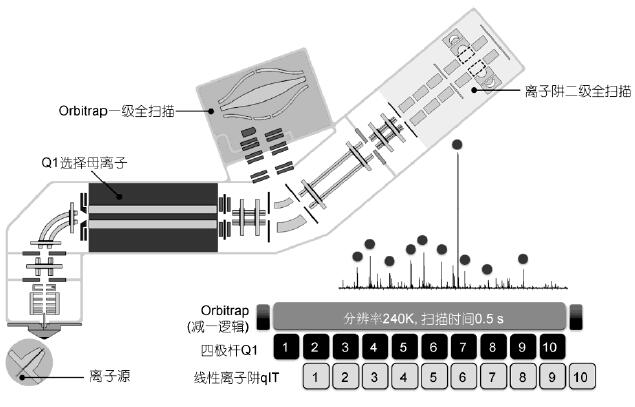
The three-in-one structure of Q-OT-qIT realizes the parallel operation that is difficult to achieve by traditional mass spectrometry, that is, simultaneous primary and secondary scanning, ion implantation, selection, fragmentation, and detection simultaneously (Fig. 7). Dynamic Scanning Management Technology Automatic control and real-time optimization of instrument operation, allowing the three mass analyzers to work together and simultaneously, minimizing the waiting time of the scanning process and the loop gap. “Subtract one logic†associates the secondary scan with the last level one scan. There is no need to wait for this first-level scan to determine the parent ion and charge, and improve the instrument running speed and analysis efficiency. Take top 10 data-dependent scanning (DDA) as an example, at 240k resolution, when the first level full scan is completed, all the second level Scanning is also done at the same time, the primary and secondary levels are uncorrelated and independent (Figure 7). In addition, the top-speed mode stimulates the secondary scan as much as possible in a fixed time, which is more than the traditional Top-N mode. Collecting low-abundance peptides also ensures the number of first-order scan points, which is conducive to simultaneous qualitative and quantitative.
The experimental results show that 1 φg of HeLa sample collected 53209 spectra in a 50-minute effective gradient with a scanning speed of 17.7 Hz, including a 120k resolution first-order orbitrap scan. The parallel operation mode improves the overall operating speed of the instrument. Under the strict card value (1% FDR), 20860 non-redundant peptides and 3865 non-redundant proteins were identified, and the latest literature reports that at least 2~3h is required to achieve the same result [9]. The number of proteins identified by Hebert et al. [6], but the amount of protein is lower, and not repeated, is more than 2 times higher than the reported results of other instruments [8,9]. In terms of scanning speed, Q- Although OT-qIT does not reach the limit speed of Q-TOF mass spectrometry, the spectral quality is higher, and the spectral resolution is 2~3 times higher than that of Q-TOF mass spectrometry [8].
The resolution of the spectrum of the 1 μg HeLa sample in many experiments was basically over 60%, and even reached 83.1% (Table 3) in the case of increasing the ion implantation time and the number of injections, showing good spectral quality. The sample system is complex. Under the short gradient, there will be interference between the co-eluting peptide and matrix impurities. The secondary response of a large number of low-abundance peptides is also weak. In this case, the resolution of the spectrum is over 80%, which is significantly higher. In the current literature [8,9]. In addition, the sample size determines the number of protein identification, traditional proteomic analysis requires micrograms of whole protein, and this experiment attempts to reduce the sample size to the nanogram, ie 50 ng The sample size has reached the current level of micrograms, providing a possibility for low concentration samples and very low abundance protein analysis.
The three-in-one structure of Q-OT-qIT makes the scanning method flexible. The multi-pole ion channel between the C-trap and the ion trap allows the ions to move freely in the forward and reverse directions, realizing the CID/HCD/ETD three fragmentation modes. MSn is used at any level, and any orbitrap or ion trap detection is used arbitrarily to produce a variety of combined scan modes. Among them, CID fragmentation is slightly more sensitive than HCD, but the speed is slightly slower and has a one-third effect, ie Low-mass-end discrimination, while HCD has abundant fragmentation information in the low-molecular-weight segment; the ultra-high resolution and mass accuracy of the orbitrap can minimize interference, while the ion trap is faster and can operate simultaneously with the orbital well, improving Efficiency. It can be seen that due to different mechanisms, various scanning modes have different effects and have certain complementarity. Q-OT-qIT's ability to control ion implantation time and injection quantity is also an important factor to ensure analysis efficiency and sensitivity. The longer the number of injections, the higher the sensitivity and the higher the spectral quality, but the slower the scanning speed. Conversely, the smaller the number of ion implantation, the faster the speed, but the lower the abundance ion loss, the lower the sensitivity. It can be seen that the experimental purpose and sample are different, and the suitable scanning modes and parameters are different. Therefore, the experiment further investigates the influence of scanning mode and ion implantation parameters on the results.
The results show that different fragmentation modes and detection methods have good complementarity. 1/2 of the identification results between the orbitrap and the ion trap are not repeated, and 2/3 results between HCD and CID are not repeated, different scans. The combination of methods can achieve greater coverage. On the other hand, the linear ion trap has higher mass accuracy than the traditional ion trap, and the mass deviation is stable within 0.2 Da, reaching the medium-high resolution mass spectrometer level. Therefore, both OT and IT scans have Higher spectral resolution. In terms of ion control, by increasing the maximum injection time and the maximum number of injections, the amount of ion implantation can be increased, and the sensitivity of low-concentration samples and extremely low-abundance proteins can be increased. The analysis speed and the number of identification of conventional samples. Through the investigation and optimization of several models and parameters, it provides more options for rapid analysis and deep coverage of proteomics.
4 Conclusion
In this paper, Q-OT-qIT three-in-one mass spectrometry was used to rapidly identify HeLa and yeast whole proteins by short gradient: (i) 50 min effective gradient, 20860 non-redundant peptides and 3865 non-redundant from 1 μg HeLa The remaining protein, the time is reduced by 50%~60% compared with the current report; (ii) 50 ng very low sample size, 14100 non-redundant peptides and 2877 non-redundant proteins were identified, and the spectral resolution rate exceeded 50%. (iii) Investigating the similarities and differences between HCD/CID fragmentation mode and IT/OT detection methods, the results show that the different methods have good complementarity and have higher spectral resolution; (iv) Investigate the maximum ion implantation The effect of time and automatic gain control on the results indicates that increasing the injection time and the number of injections can significantly improve the detection sensitivity of low-concentration samples. In summary, based on Q-OT-qIT mass spectrometry, this paper realizes the complex sample protein in a short time. Group depth coverage analysis, the "one hour proteome" era has arrived.
Figure 7 Q-OT-qIT mass spectrometry structure and parallel operation principle
At the same time as the full scan of the orbital well, the quadrupole is sequentially selected for several parent ions, and the ion trap sequentially scans the secondary fragments of several parent ions, and the three are simultaneously operated.
Acknowledgments Some of the image material was provided by the Thermo Fisher Scientific San Jose plant.
references
1 Delahunty CM, Yates III J R. MudPIT: multidimensional protein identification technology. Biotechniques, 2007, 43: 563–569
2 Jones KA, Kim PD, Patel BB, et al. Immunodepletion plasma proteomics by triple TOF 5600 and orbitrap elite/LTQ-orbitrap Velos/Q exactive mass spectrometers. J Proteome Res, 2013, 12: 4351–4365
3 Mann M, Kelleher N L. Precision proteomics: the case for high resolution and high mass accuracy. Proc Natl Acad Sci USA, 2008, 105: 18132–18138
4 Geiger T, Wehner A, Schaab C, etal. Comparative proteomic analysis of eleven common cell lines reveals ubiquitous but varying expression of most proteins. Mol Cell Proteomics, 2012, 11: M111.014050
5 Senko MW, Remes PM, Canterbury JD, et al. Novel parallelized quadrupole/linear ion trap/orbitrap tribrid mass spectrometer improving proteome coverage and peptide identification rates. Anal Chem, 2013, 85: 11710–11714
6 Hebert AS, Richards AL, Bailey DJ, et al. The one hour yeast proteome. Mol Cell Proteomics, 2013: M113.034769
7 Nagaraj N, Kulak NA, Cox J, et al. System-wide perturbation analysis with nearly complete coverage of the yeast proteome by single-shot ultra HPLC runs on a bench top orbitrap. Mol Cell Proteomics, 2012, 11: M111.013722
8 Andrews GL, Simons BL, Young JB, et al. Performance characteristics of a new hybrid quadrupole time-of-flight tandem mass spectrometer (TripleTOF 5600). Anal Chem, 2011, 83: 5442–5446
9 Kelstrup CD, Young C, Lavallee, R, et al. Optimized fast and sensitive acquisition methods for shotgun proteomics on a quadrupole orbitrap mass spectrometer. J Proteome Res, 2012, 11: 3487–3497
The One Hour Proteome: Q-OT-qIT Mass Spectrometry Based Rapid Proteome Identification
ZHANG Wei, GU PeiMing, JIANG Zheng, MING Hong & CHEN Wei
ThermoFisher Scientific China, Shanghai 201206, China
With the realization of "the one hour proteome", in-deep proteome under rapid LC-MS/MS gradient is achieved. The current study utilized novel Q-OT-qIT mass spectrometer to analyze and optimize "the one hour proteome". Within 50 min LC-MS/MS gradient, a total of 20860 and 14100 unique peptides were identified from 1 μg and 50 ng HeLa digest, corresponding to 3865 and 2877 proteins, respectively. Similar result from current reports used at least 2–3 h The effects of various collision modes, detectors, maximum injection times and automatic gain control settings are further evaluated, and the complementarity between different acquisition methods and scan parameters were described. further, the massive parallelization of Q-OT-qIT was discussed, Provide useful information for rapid proteome analysis.
Proteomics, rapid identification, deep coverage, orbitrap, Q-OT-qIT MS
Doi: 10.1360/10.1360/052014-16
Next Article
Medicinal primer, exactly what use?